Example Output¶
Now that you have completed your alignment based profiling using MiFoDB, we can calculate the mapping abundance.
Table setup¶
1. Your inStrain profile results
genome |
coverage |
breadth |
nucl_diversity |
length |
true_scaffolds |
detected_scaffolds |
coverage_median |
coverage_std |
coverage_SEM |
breadth_minCov |
breadth_expected |
nucl_diversity_rarefied |
conANI_reference |
popANI_reference |
iRep |
iRep_GC_corrected |
linked_SNV_count |
SNV_distance_mean |
r2_mean |
d_prime_mean |
consensus_divergent_sites |
population_divergent_sites |
SNS_count |
SNV_count |
filtered_read_pair_count |
reads_unfiltered_pairs |
reads_mean_PID |
reads_unfiltered_reads |
divergent_site_count |
C-03.Ssa-BR.fna |
1.686020547 |
0.049164091 |
0.004595774 |
1896140 |
182 |
86 |
0 |
69.19478668 |
0.050739639 |
0.011300326 |
0.774346839 |
0.000140703 |
0.986372334 |
0.988145797 |
FALSE |
242 |
39.69008264 |
0.951699521 |
0.999845137 |
292 |
254 |
252 |
165 |
15171 |
15417 |
0.981642137 |
36199 |
417 |
|
EBC_086.5.fna |
1.596317454 |
0.049848898 |
0.006035971 |
2377866 |
79 |
52 |
0 |
19.94120243 |
0.012974942 |
0.028909535 |
0.755746415 |
0.002048653 |
0.979081506 |
0.984682077 |
FALSE |
1337 |
56.69334331 |
0.637899652 |
0.9941014 |
1438 |
1053 |
1040 |
825 |
17829 |
19210 |
0.969968582 |
48221 |
1865 |
2. Sample read info, found in bowtie2.log file created after making the .bam file. For each bowtie2.log, save the sample name and paired reads (in this example 18233183 before (100.00%) were paired, which is the read_pairs after adapter trimming and human genome remover) .. code-block:
$ head bowtie2.EBC_087.log
18233183 reads; of these:
18233183 (100.00%) were paired; of these:
16282298 (89.30%) aligned concordantly 0 times
1046019 (5.74%) aligned concordantly exactly 1 time
904866 (4.96%) aligned concordantly >1 times
----
16282298 pairs aligned concordantly 0 times; of these:
520393 (3.20%) aligned discordantly 1 time
----
15761905 pairs aligned 0 times concordantly or discordantly; of these:
3. Database mapping file MiFoDB_beta_v2_allRef
Calculate relative abundance:¶
1. Join the IS_genome_info.csv file to sample read info and sample mapping information.
percent_abundance = ((filtered_read_pair_count)/read_pairs)*100))
Where filtered_read_pair_count is originally in the .IS_genome_info.csv, and read_pairs is from bowtie2.log
It should look something like this:
genome |
sample |
length |
coverage |
abundance |
breadth |
filtered_read_pair_count |
read_pairs |
EBC_086.5 |
EBC_087 |
2377866 |
2.03925873030692 |
0.215016678311685 |
0.150023592582593 |
38578 |
17941864 |
GCF_001039045.1_ASM103904v1_genomic |
EBC_087 |
2899876 |
1.27013224013716 |
0.147297961906299 |
0.019880160393065 |
26428 |
17941864 |
GCF_001434915.1_ASM143491v1_genomic |
EBC_087 |
2232918 |
0.739709653466898 |
0.0614707591139917 |
0.0044753098859877 |
11029 |
17941864 |
GCF_002276885.1_ASM227688v1_genomic |
EBC_087 |
2495148 |
2.08628466127059 |
0.218199179304893 |
0.0112313978970385 |
39149 |
17941864 |
GCF_003641185.1_ASM364118v1_genomic |
EBC_087 |
3671373 |
1.62835157310358 |
0.244311293408533 |
0.0552324702502306 |
43834 |
17941864 |
2. For QC, filter any genomes with breadth < 0.5. Those can be considered “low confidence” mapping, while any genomes with breadth > 0.5 are considered high-confidence mapping results.
You can then combine all results from MiFoDB_prok, MiFoDB_euk, and MiFoDB_sub.
For an additional QC with MiFoDB_sub, remove any genome with abundance <2%.
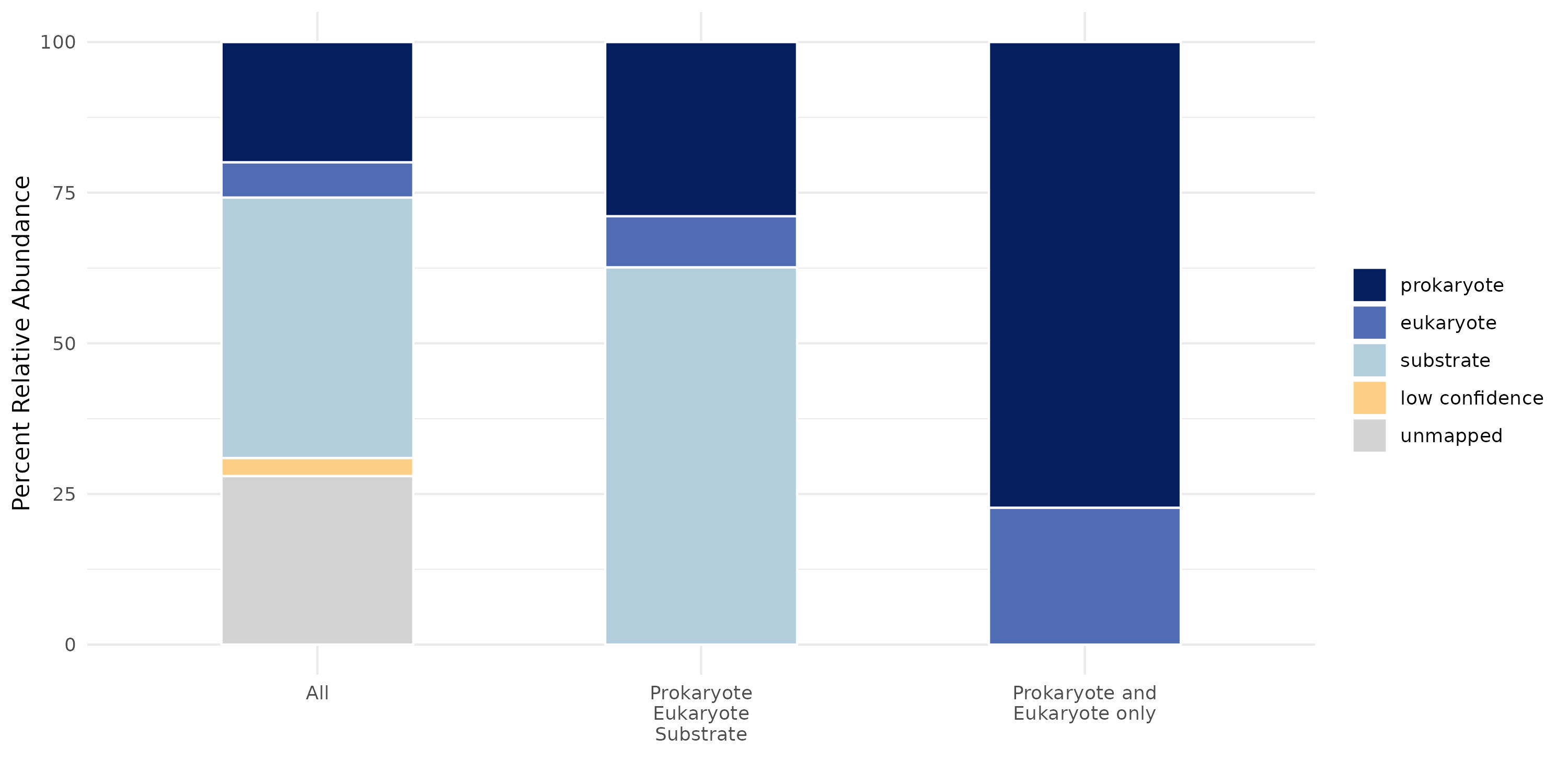
3. Results are now ready for plotting and downstream analysis. For example:
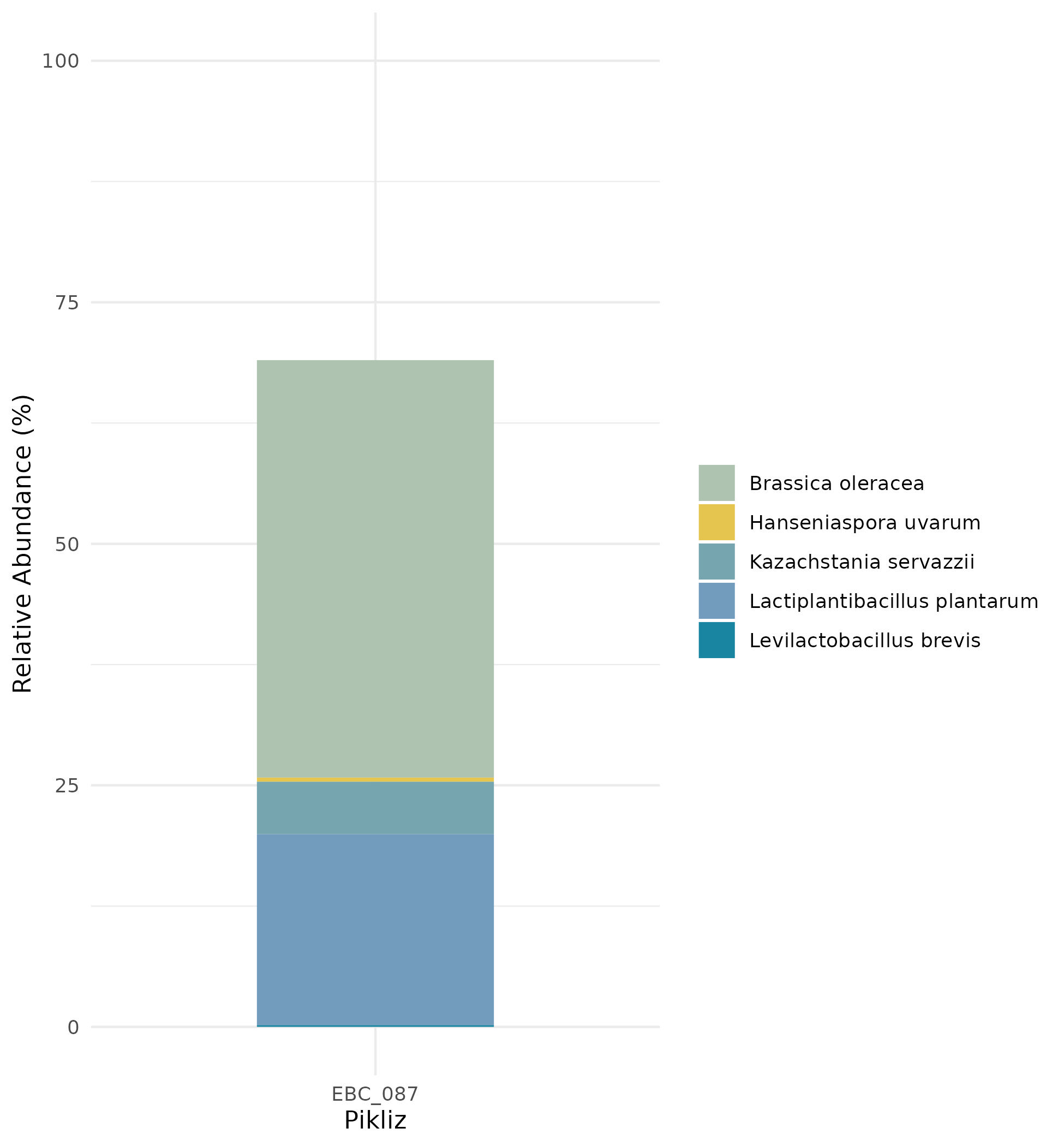
Or take a closer look at the mapped species: